The Rise of Room-Temperature Superconductors: Are we ready for it?
The Rise of Room-Temperature Superconductors: Are we ready for it?
The Rise of Room-Temperature Superconductors: Are we ready for it?
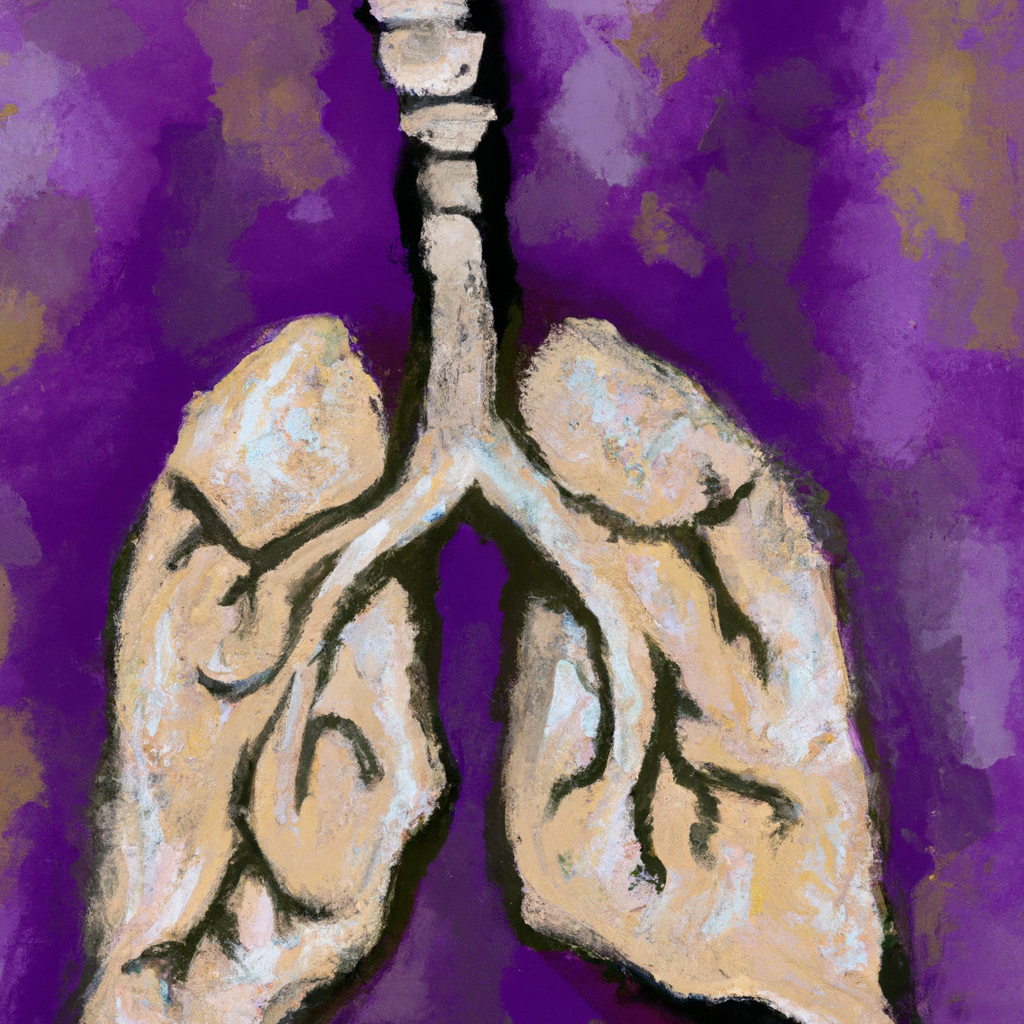
Idiopathic Pulmonary Fibrosis (IPF) is an unceasing and lethal lung disease characterized by fibrosis - scarring of the lungs. Approximately 58,000 new US cases occur annually, and the median survival timeframe post-diagnosis is just 3-5 years (source: American Lung Association). This severe illness is 'idiopathic,' an intriguing moniker indicating the lack of clarity about its causes. Despite significant research breakthroughs and the discovery of biomarkers, the true etiology of IPF remains unknown, further intensifying the struggle to determine who is predisposed to acquire the condition.
Biomarkers are biological indicators present in bodily fluids or tissues, providing essential information about physiological, pathological, and therapeutic responses. In IPF, biomarkers have been identified that correlate with disease severity, progression, prognosis, response to therapy, and mortality (source: Biomarkers in idiopathic pulmonary fibrosis). This range of biomarkers, including KL-6, SP-A, SP-D, CCL18, MMPs, and more, has enhanced understanding and aided diagnosis. However, their role within the context of cause and progression remains largely uncertain. Although an association has been observed, the intricacy of IPF's pathogenesis signifies that biomarkers alone fail to provide a complete explanation for the onset of the disease.
The "idiopathic" component of IPF implies the disease's cryptic origin. Despite several risk factors and genetic determinants being identified - such as cigarette smoking, certain viral infections, gastroesophageal reflux disease, environmental pollutant exposure, and family history - the exact interaction and influence of these factors in contributing to the development of IPF hasn't been deciphered fully (source: European Respiratory Journal). Therefore, people exhibiting identical risk factors may present drastically diverse susceptibilities to the disease, further highlighting our limited understanding of IPF's pathogenesis.

Click here to see our updated list of Pulmonary Fibrosis Antibodies
If you are interested in learning more about Pulmonary Fibrosis or looking for resources, check out the Pulmonary Fibrosis Foundation.
The next steps for IPF research involve a multi-dimensional approach, integrating combinations of genetic, environmental, and immunological factors to predict who's most likely to contract the disease. Advanced genomic techniques such as whole-genome sequencing may offer insights into the genetic makeup's role in IPF susceptibility (Source: European Respiratory Journal). Additionally, continual assessment and refinement of existing and novel biomarkers by considering epigenetic modifications, microbiome changes, and other influencing elements will be vital. Specific attention must also be paid to lifestyle components and their interaction with genetic factors.
Hopefully, the continued study of high-risk populations, ongoing medical trials, progressive genetic analysis, and a broader understanding of the disease's pathophysiology will unveil the idiopathic nature of IPF. Combining these various facets, researchers can strive towards a more definitive prediction model for the likelihood of contracting IPF, ensuring earlier diagnosis and therapeutic intervention opportunities.

Sources:
1. American Lung Association
2. Biomarkers in idiopathic pulmonary fibrosis
3. European Respiratory Journal
4. European Respiratory Journal
The Rise of Room-Temperature Superconductors: Are we ready for it?
Epigenetics, Fear Development
Asthmatics Warned About Cooking - Sensationalism in Academic Publications